We have developed methods and written code for automated reaction mechanism generation of complex systems, which can be applied to a wide range of chemistries.
Property Estimation
Computational chemistry and statistical thermodynamic calculations are often necessary to estimate kinetic rate parameters for accurate models and predictions.
Microkinetic Modeling
Conversion, yields, and selectivities are obtained by coupling the reaction mechanism with reactor design equations and solving the resultant system of equations.
Research areas
Shale Gas Conversion: CISTAR
Design a transformative engineered system to convert light hydrocarbons from shale resources to chemicals and transportation fuels in smaller, modular, local, and highly networked processing plants
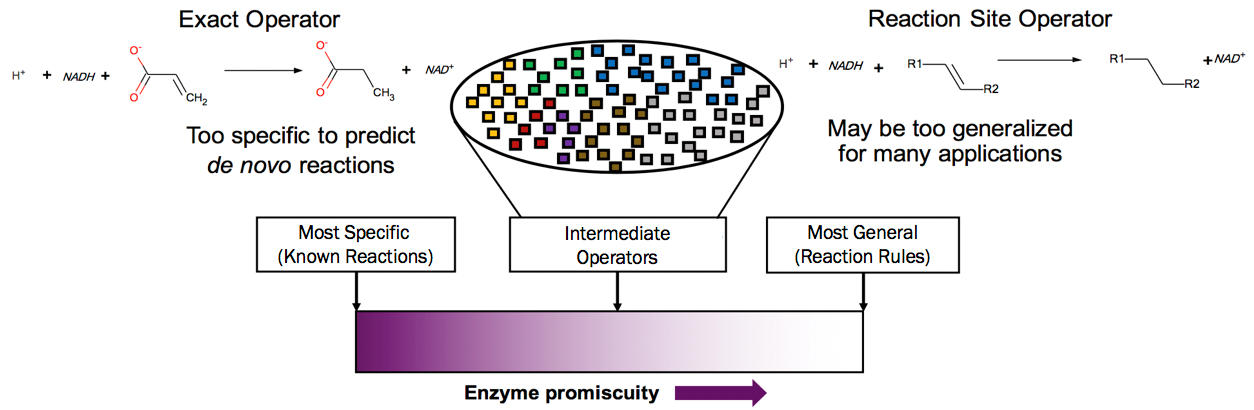
Automated network generation that defines and implements chemistry of generalized enzyme functions can provide insight into novel pathways to synthsize difficult to obtain biochemicals
Modeling of biomass fast pyrolysis, including computational lignin structure generation and catalytic upgrading of vapor-phase components of bio-oil using acidic zeolites
Develop computational approaches to understand the mechanisms contributing to property degradation and to visual changes in materials - with overall aims to preserve artwork
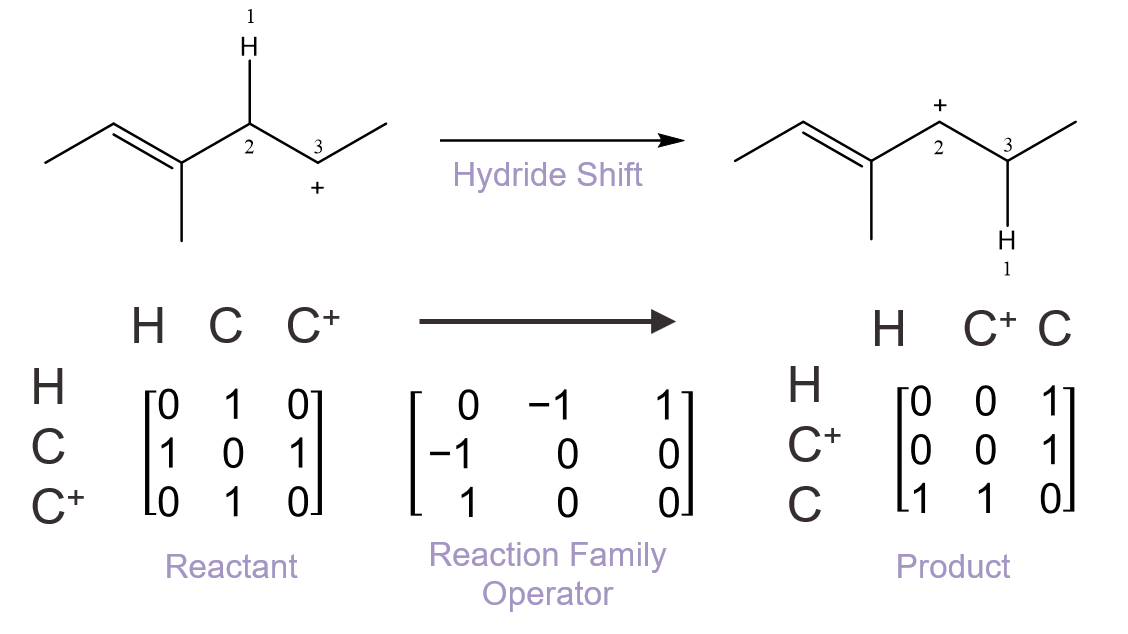
We have developed a method for automated generation of complex reaction mechanisms that allow kinetic models with great detail to be built. For our generation, molecules are represented as molecular graphs or matrices, and manipulations of these matrices allow reactions to be carried out, molecule uniqueness to be determined, and chemical/thermodynamic properties to be calculated. Reaction chemistry is organized into reaction families and a mathematical matrix operator is specified for each family. If a reaction is found to be possible, the program executes the mathematical transformation via matrix addition and the new connectivity for the products is determined. Unique matrix products are appended to the unreacted species list that is searched until all combinations of species and reaction families have been examined. The automatically generated network can then be integrated with our other in-lab codes that automatically solve the network, such as applying reactor design equations and integrating in a microkinetic model. We have applied our methodology to a wide range of different problems, including production of silicon nanoparticles, biochemical transformations, polymerization and depolymerization, and tropospheric ozone formation.
Property Estimation
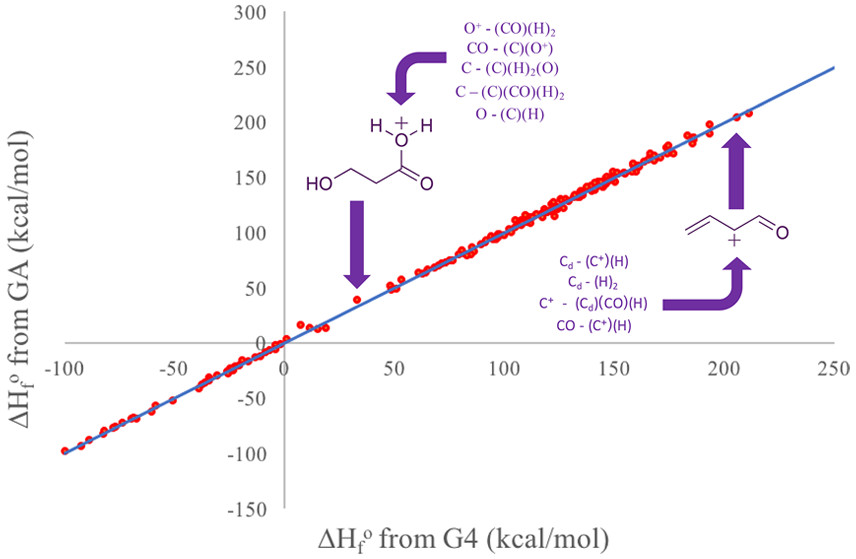
Reaction coefficients are calculated a variety of ways, often tailored for each reaction situation. Most reactions are described through the Arrhenius relationship, but both the network generation and microkinetic modeling tools are flexible to different representations. Pre-exponential factors and activation energies can likewise be calculated through various ways, including Bell-Evans-Polanyi, transition state theory, statistical thermodynamics, or density functional theory. These calculated values can be used in the microkinetic model to determine rates throughout the reactor.
Microkinetic Modeling
Microkinetic modeling is a powerful approach for studying complex catalytic systems. In a microkinetic analysis, all elementary steps comprising the reaction mechanism are considered explicitly, and no assumptions about rate-determining steps are made. For our microkinetic models, we combine our methodology for computer generations of reaction mechanisms as well as our calculations for kinetic parameters. Reaction chemistry is organized into reaction families and a mathematical matrix operator is specified for each family. The reaction mechanism is created automatically by applying the operators to all the different reactants and their products. Structure / reactivity relationships, one for each reaction family, are used to specify the rate coefficients. Rate coefficients are calculated through quantum chemistry, statistical mechanics, or density functional theory to name a few. Reactor design equations are setup with the reaction mechanism and rate coefficients, and the systems of equations are solved to determine concentration profiles and reaction rates of all species throughout the reactor. The results of microkinetic modeling can tell us important properties of a system, such as the most predominant species exiting the reactor and on the surface of the catalyst, overall turn-over frequency as a measure of catalytic activity, and selectivity of reaction system towards a desired product. Additionally, we can probe the sensitivity of the model to individual model parameters, such as rate constants, equilibrium coefficients, or binding energies.
Shale Gas Conversion: CISTAR, Center for Innovative and Strategic Transformation of Alkane Resources
Since the Shale Gas Revolution in 2008, hydraulic fracturing has unlocked vast quantities of hydrocarbons that were previously unavailable, greatly expanding the supply of recoverable unconventional shale gas. With this revolution brings the potential to lower the cost of our nation’s electricity, power, chemicals, and fuels for the next 100 years with this newly recoverable natural gas. However, shale gas is compromised of more than just methane / natural gas. Light alkenes are a significant weight fraction of recovered hydrocarbons. The US Energy Information Administration (EIA) projects the US natural gas production will increase through 2050, growing an estimated 59% from 2017 to 2050; correspondingly, this means a significant increase in light hydrocarbon recovery. Production of these hydrogen-rich hydrocarbon resources is expected to grow at a rate 40% higher than the production of unconventional oil. However, there is a significant gap in our nation’s ability to effectively upgrade and use these rich light hydrocarbon reserves for petrochemicals and fuels.
CISTAR aims to use the surplus of light alkenes from shale gas more efficiently, by dehydrating and oligomerizing short alkanes to liquid fuels in small module units close to the wellhead. Effective production of chemical-grade feedstocks and clean fuels close to reserve sources will reduce the need to transport hydrocarbons long distances by rail, highway, or pipeline. Such transformative changes in hydrocarbon processing will vastly lower costs, reduce production emissions, improve safety, and sustain the wealth of national fuel that can “bridge: to next-generation sustainable energy.
Our lab focuses on modeling and optimizing the olefin oligomerization reactions to higher molecular weight products for various catalyst activities, reaction networks, and product distributions. We employ kinetic Monte Carlo to better understand surface interactions with catalysts as well as reaction network generation and reactor integration for tandem catalytic pathways. In line with CISTAR’s goals, we aim to described experimental results or suggest new catalytic material through the control of catalytic chemistry, finding the most desirable molecular weight distribution that can be condensed and easily transported for fuel use.
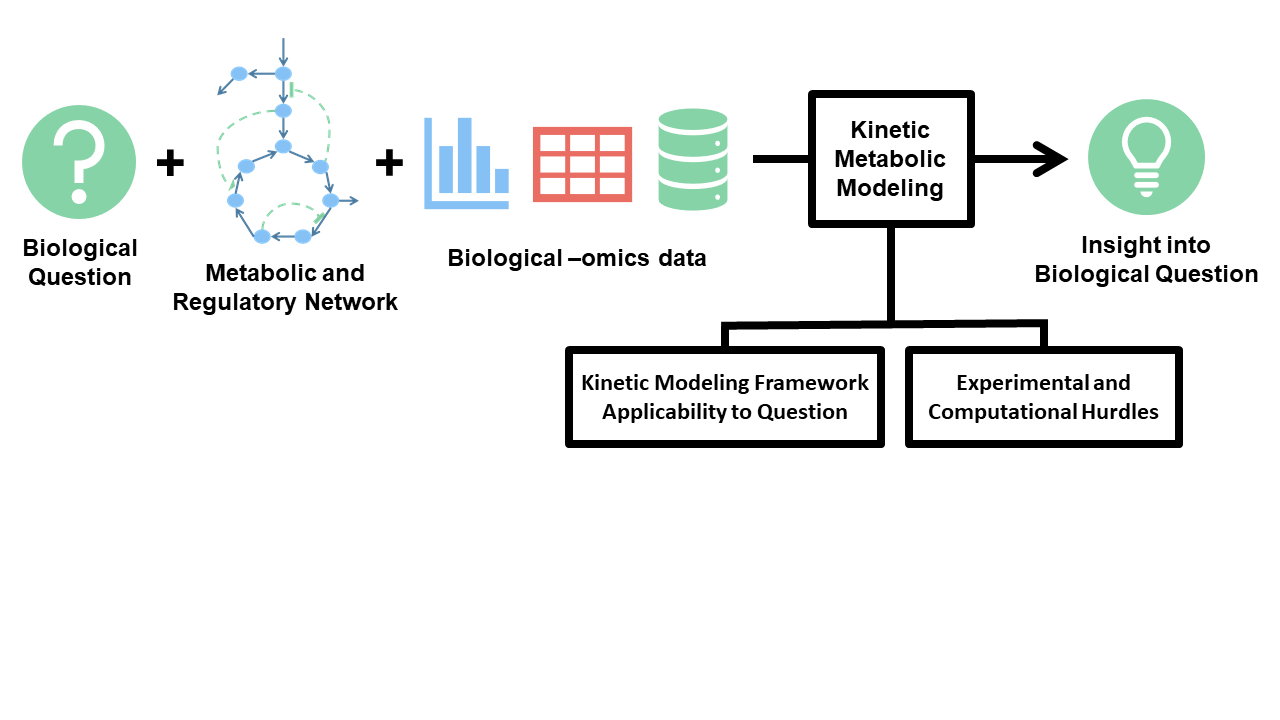
Growing energy consumption, finite petroleum resources, combined with current state of climate changes, have all pointed to the need for developing more sustainable and renewable industrial processes. Microorganisms can be engineered into microbial factories to take in waste biomass such as syngas and lignin, and readily produce valuable chemicals ranging from biofuels, commodity chemicals, to pharmaceuticals. The Broadbelt Lab, in collaboration with the Tyo Lab, is developing computational frameworks to guide and optimize metabolic network design to achieve production of desirable compounds in biological systems. These efforts are broadly directed in two main areas, which are predicting enzyme promiscuity for novel biochemistry, and in developing kinetic models of metabolism.
We are developing a platform to explore de-novo enzymatic reactions leading to diverse compounds, which may not have known associated biological routes. Many enzymes, along with their primary activities, possess capabilities of catalyzing a side reaction with similar mechanisms but different substrates. The extent of enzyme promiscuity is not fully studied, thus representing enormous possibilities for biosynthesis of non-native compounds. By compiling generalized enzyme functions based on database of known metabolic pathways, we can create enzymatic operators, or mathematical representation of reaction mechanisms, which are then iteratively applied to a metabolite to generate new compounds. Subsequently, by grouping operators based on reaction similarity, and fine-tuning levels of operator specificity, we can prune the operators to only include biologically realistic levels of novelty. Our cheminformatics approach accelerates the identification of promising metabolic routes and candidate enzyme information, to guide and inform future experimental efforts.
Beyond individual reaction pathways, the complexity of metabolic networks presents further issues for the goal of creating useful biochemicals. Biochemical networks are highly interconnected and include multiple layers of regulation, and so the ideal engineering strategy to improve production of a biochemical target is rarely obvious. Toward this end, we have developed kinetic models of metabolic networks, which allow us to build mechanistically-derived, ODE-based models which can predict the effects of typical metabolic engineering efforts, such as changes to metabolite or enzyme levels, in order to suggest engineering targets. Nonetheless, these systems have many more parameters than can be identified exactly through experimental means, and therefore we have utilized multiple approaches toward this problem of parameter inference. One such strategy is ensemble modelling, in which multiple parameter sets are generated in agreement with a reference state and then screened against perturbation states. Another approach we have taken has been to combine approximate rate law forms and techniques which identify parameters using Bayesian inference.
By drawing inspiration from other modelling fields, we have also developed several state-of-the-art strategies for accelerating kinetic model performance, including identifying conserved species, checking systems for local stability, and sorting parameter screening order. Currently, we are researching new modelling frameworks which will allow us to translate these metabolic models into other organisms and metabolic pathways, as well as utilize a wide range of experimental data types.
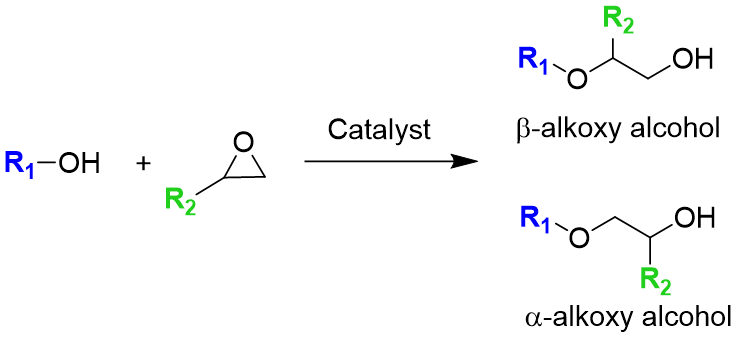
Heterogeneous and Homogeneous Catalysis: Polyol Synthesis from Epoxides
Polyol resins are heavy molecular weight, highly crosslinked polymers with hydroxyl functionality, with uses spanning polyurethane and surface coating industries. The epoxide ring-opening reaction is a widepresad method for functionalizing chemical chains, especially for vegetable oils. Most commonly, the synthesis of polyol from vegetable oils comprises of a two-step synthesis via epoxidation and alcoholysis.
Selective ring-opening of epoxides could lead to a number of useful products or synthons, providing a useful probe reaction for understanding acid catalysts. Collaborating with the Notestein lab as well as Dow, we use computational techniques and
experimental data to develop homogeneous and hetergeneous regioselective catalysts and optimize reaction conditions.
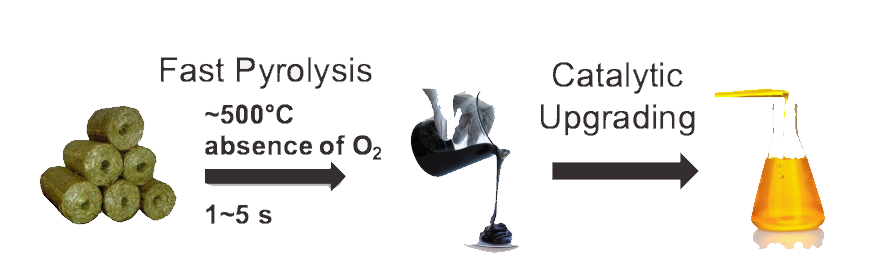
Lignocellulosic biomass has the potential to serve as the renewable resource that propels the US towards its sustainable and green energy goals. To date, lignin has mainly been utilized as low value fuel for process heat; however, the aromatic nature and low oxygen-to-carbon ratio of lignin provide the potential for production of specialty fuels and chemicals. heat. One of the roadblocks to effectively converting lignin into usable fuels is that the structure of lignin has yet to be entirely understood owing to its polydispersity, complexity, and hyper-branched topology. Libraries of structural representations of lignin accounting for these facets have recently been proposed for wheat-straw based on a stochastic generation method that creates lignin molecules that collectively conform to properties measured experimentally. We have extended this stochastic method to accommodate more complexity and any type of biomass (i.e., softwood, hardwood, or herbaceous). The unique mechanistic details for several of the new lignin bond types are essential in deciding rules for bond formation in the algorithm.
Further, methods of decreasing the degrees of freedom during optimization of crucial parameters has been investigated. Apart from generating libraries of lignin structures, the added complexity allows for the exploration of “lignin space”, which can represent all possible structures of lignin given the experimental characteristics of monomer distribution, bond distribution, molecular weight distribution, and branching coefficient. Thus, we aim to create lignin libraries for any biomass source with reliable and consistent experimental data can be generated for future kinetic modeling studies or molecular simulations.
CuBISM is at the intersection of art and science, focusing on art as a catalyst for the development of innovative and holistic approaches to change over time in complex material systems. Computational tools are used to better understand how and why artistic materials degrade; a central challenge is tieing the visual appearance of the painted surfaces to the underlying molecular and microscopic material structures. The analystical and methodological tools developed by the project will have broad relevance to the materials science and conservation science communities, illustrating the creative aspects of both science and art.
The drying and oxidative degradtion of linseed oil is especially challenging. The oxidative degradation of linseed oil consists of the continuation of the hardening process and the oxidation takes place on the alkylic segments, leading to partial fragmentation of the structure. Models for these complex oils not only have to handle fast reactions occuring in microseconds, but also long-term molecular chain changes over decades.





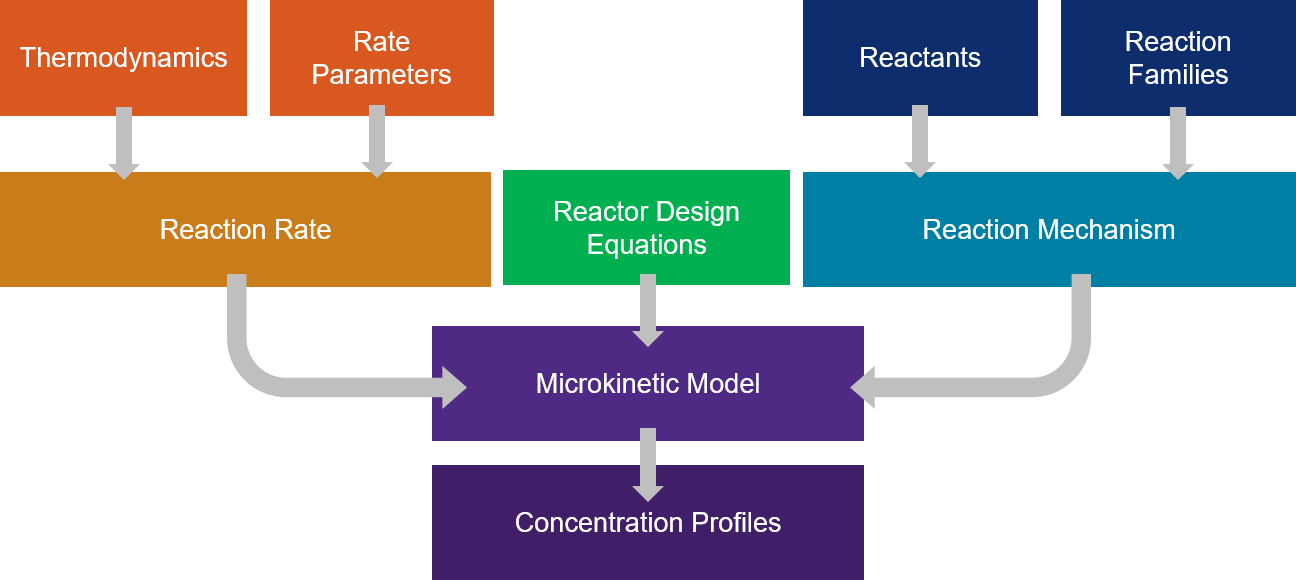 Microkinetic modeling is a powerful approach for studying complex catalytic systems. In a microkinetic analysis, all elementary steps comprising the reaction mechanism are considered explicitly, and no assumptions about rate-determining steps are made. For our microkinetic models, we combine our methodology for computer generations of reaction mechanisms as well as our calculations for kinetic parameters. Reaction chemistry is organized into reaction families and a mathematical matrix operator is specified for each family. The reaction mechanism is created automatically by applying the operators to all the different reactants and their products. Structure / reactivity relationships, one for each reaction family, are used to specify the rate coefficients. Rate coefficients are calculated through quantum chemistry, statistical mechanics, or density functional theory to name a few. Reactor design equations are setup with the reaction mechanism and rate coefficients, and the systems of equations are solved to determine concentration profiles and reaction rates of all species throughout the reactor. The results of microkinetic modeling can tell us important properties of a system, such as the most predominant species exiting the reactor and on the surface of the catalyst, overall turn-over frequency as a measure of catalytic activity, and selectivity of reaction system towards a desired product. Additionally, we can probe the sensitivity of the model to individual model parameters, such as rate constants, equilibrium coefficients, or binding energies.
Microkinetic modeling is a powerful approach for studying complex catalytic systems. In a microkinetic analysis, all elementary steps comprising the reaction mechanism are considered explicitly, and no assumptions about rate-determining steps are made. For our microkinetic models, we combine our methodology for computer generations of reaction mechanisms as well as our calculations for kinetic parameters. Reaction chemistry is organized into reaction families and a mathematical matrix operator is specified for each family. The reaction mechanism is created automatically by applying the operators to all the different reactants and their products. Structure / reactivity relationships, one for each reaction family, are used to specify the rate coefficients. Rate coefficients are calculated through quantum chemistry, statistical mechanics, or density functional theory to name a few. Reactor design equations are setup with the reaction mechanism and rate coefficients, and the systems of equations are solved to determine concentration profiles and reaction rates of all species throughout the reactor. The results of microkinetic modeling can tell us important properties of a system, such as the most predominant species exiting the reactor and on the surface of the catalyst, overall turn-over frequency as a measure of catalytic activity, and selectivity of reaction system towards a desired product. Additionally, we can probe the sensitivity of the model to individual model parameters, such as rate constants, equilibrium coefficients, or binding energies.